

女,34岁
主诉:进行性言语表达不畅10月余,加重1周
实验室检查:无特殊
肌电图:进行性和慢性失神经表现
病案讨论:

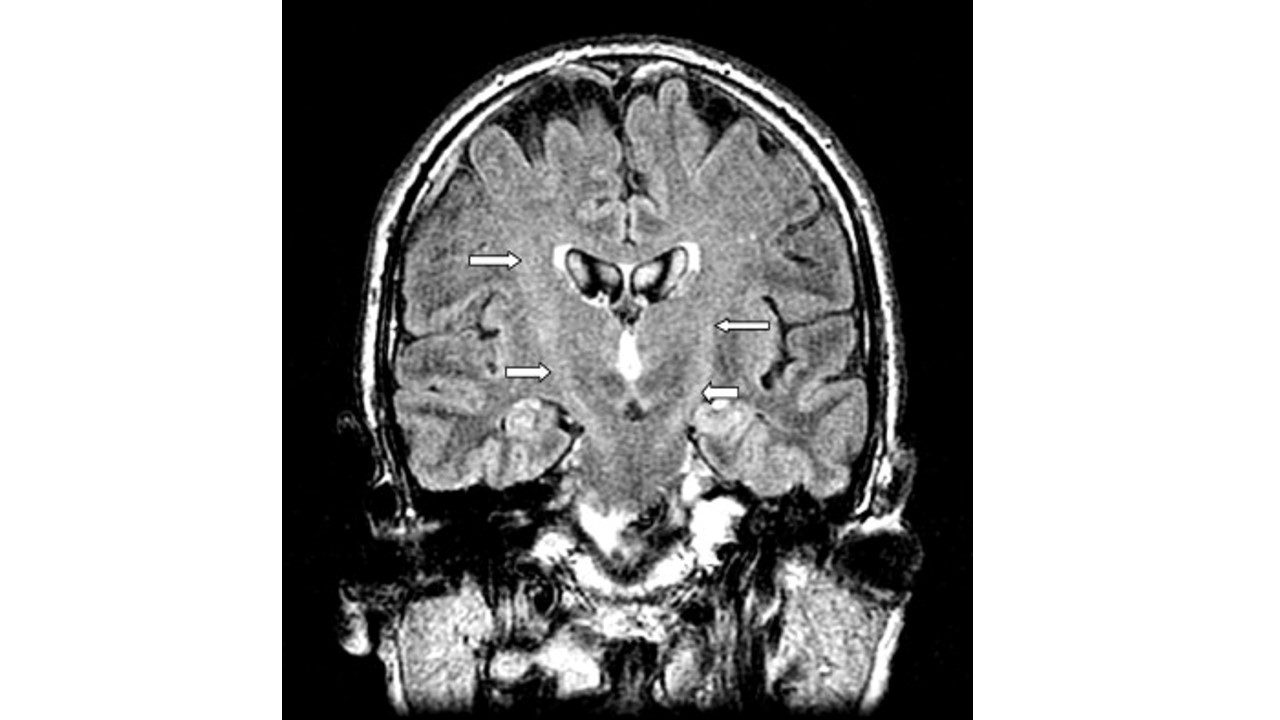
影像表现:中央前回至脑干的双侧皮质脊髓束走行区条状 T2WI/T2-FLAIR及DWI高信号影。
临床诊断:肌萎缩侧索硬化(ALS)。
概述:肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS),即运动神经元病,俗称“渐冻症”,主要影响原发性运动皮层、脑干和脊髓的运动神经元,导致上运动神经元(upper motor neuron,UMN)、下运动神经元(lower motor neuron,LMN)受累的一种罕见的神经退行性疾病。患者多中年起病(30~60岁),男女性别比3:2,具有高致死率,无法治愈,平均生存期约2~5年;
发病机制:病因不明,可能与遗传、病毒及免疫系统功能紊乱、自由基、毒素等有关,大多为散发,5%~10%的患者有家族史,遗传方式主要为常染色体显性遗传;
临床分类:根据上、下运动神经元损害的不同组合,可分为经典型ALS、进行性肌萎缩(PMA)、进行性延髓麻痹(PBA)和原发性侧索硬化(PLS);
临床表现:具有异质性,主要表现为不同组合的肌无力、萎缩和皮质脊髓束征,其中以ALS最为典型,表现为球部、四肢、胸腹部肌肉进行性无力和萎缩。常见首发症状为一侧或双侧手指活动笨拙、无力,少数患者从下肢、舌肌受累开始。受累部位常有明显肌束颤动(肌肉跳动)、肌肉萎缩,同时伴有腱反射活跃或亢进,Hoffmann征阳性、Babinski征阳性。20%~50%的患者可以表现有认知功能障碍,5%~15%的患者甚至会发展为额颞叶痴呆。大多发病后平均3~5年因呼吸衰竭死亡;
诊断标准:目前ALS尚无可靠的生物学标志物,首发症状复杂多样、易与其他神经退行性病相混淆,故早期诊断极为困难。临床诊断仍是主流,具体参照2000年EI Escorial修订版ASL标准,结合临床、电生理及病理检查做出综合判断,应同时满足:①至少三个解剖区域(延髓、颈、胸、腰)出现上/下运动神经元受累的证据;②在一个部位内扩展或从一个部位扩展到其他部位;③排除其他疾病的干扰;
MRI检查的作用:ALS的诊断主要依靠临床症状和肌电图,MRI为首选检查方法,虽然MRI不能对ALS提供确诊依据,但能清晰显示脑内病灶部位及逐渐增多、增大趋势,有助于ALS与其他疾病的鉴别,排除结构性损害。
MRI表现:
①常规MRI早期可见沿中央前回至脑干的双侧皮质脊髓束走行区条状 T2WI/T2-FLAIR及DWI高信号,冠状位观察典型的“倒八字征”;
②部分ALS患者中央前回皮质变薄,可见中央前回皮质内线状分布的T2WI或T2*/SWI低信号,典型的“运动带征”,可能跟铁沉积有关,提示铁的沉积与进行性神经元变性有关;
③DTI:皮质脊髓束走行区FA值降低,MD值和ADC值升高;
④晚期可出现海马、脊髓等萎缩改变。
鉴别诊断:
华勒氏变性:见于各种皮质及皮质下损害,MRI表现为沿皮质脊髓束的异常信号,多为单侧受累,且皮质脊髓束的信号强度呈动态变化;
海洛因中毒:有毒物接触史,MRI表现为双侧小脑齿状核、胼胝体、皮质脊髓束对称性条片状T2WI/T2-FLAIR高信号。
Krabbeb病:由于半乳糖神经酰胺酶缺乏导致一种罕见常染色体隐性遗传病,可通过基因检测确诊,MRI除了皮质脊髓束受累,还可以出现胼胝体压部、视辐射片状T2WI/T2-FLAIR高信号。
类型:原创
病例ID:ZYLM000003017
校对:王宇军
阅读:2231
文章已于2023-09-11修改




