


男,37岁
主诉:脊髓小脑共济失调15年,加重2月;x=
现病史:脊髓小脑共济失调15年(2009年),基因诊断(2019年),近2月进展,言语含糊,留置尿管。
病案讨论
影像表现:两侧侧脑室旁、胼胝体压部、脑桥多发脑白质脱髓鞘改变,中脑、脑桥及小脑萎缩。
常染色体显性脊髓小脑性共济失调(Spinocerebellar ataxia,SCA)是最常见的常染色体显性遗传性共济失调(Autosomal Dominant Cerebellar Ataxias,ADCAs),它是一组在临床和遗传上具有高度异质性的遗传性神经变性疾病,其核心临床和神经病理学特征是小脑变性,与眼肌麻痹、锥体束征、锥体外系体征、痴呆、色素视网膜病变、癫痫发作、下运动神经元体征或周围神经病变有不同程度的相关性[1]。SCA起病年龄通常在30至50岁之间,也可出现在幼年或老年时期[2]。通常扩增等位基因上的CAG重复数越多,发病年龄越早,病情越重。目前倾向于根据遗传位点对SCA进行分类,每种亚型都被命名为SCAn(n依照致病基因或位点发现的时间顺序递进),目前已经报道了超过41种不同的SCA亚型[3]。
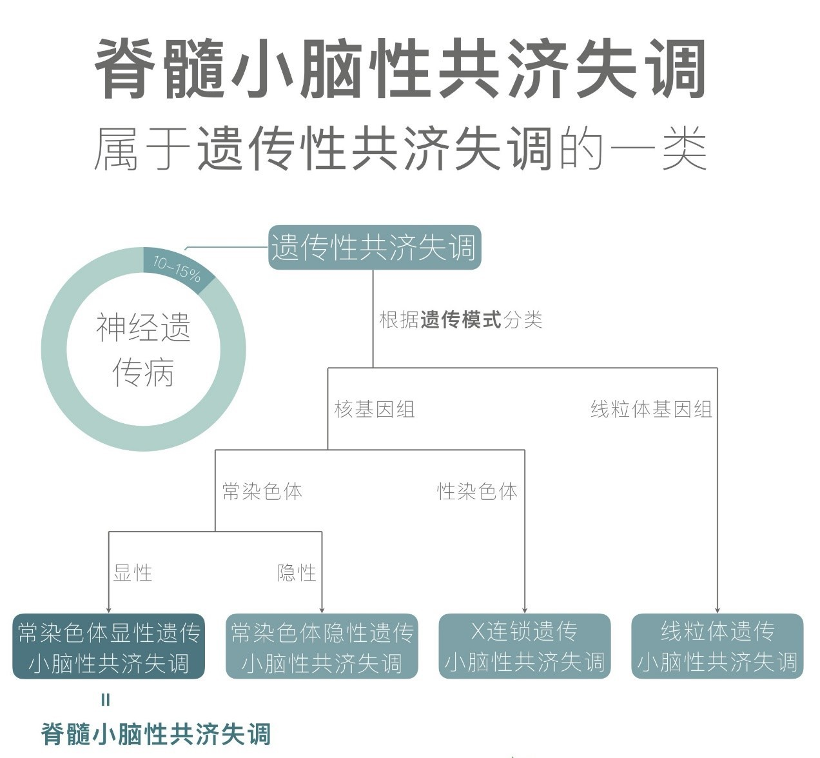
图1 SCA属于遗传性共济失调的一类
SCA亚型分类
根据基因突变的类型,可以将SCA分为两大类:
1、重复扩展性SCA,此类又可分为多聚谷酰胺SCA(由翻译区CAG动态重复突变引起,又称为翻译区重复扩展型SCA)、非翻译区重复扩展型SCA;
2、非重复扩展型SCA,由错义、缺失、插入等传统突变引起。
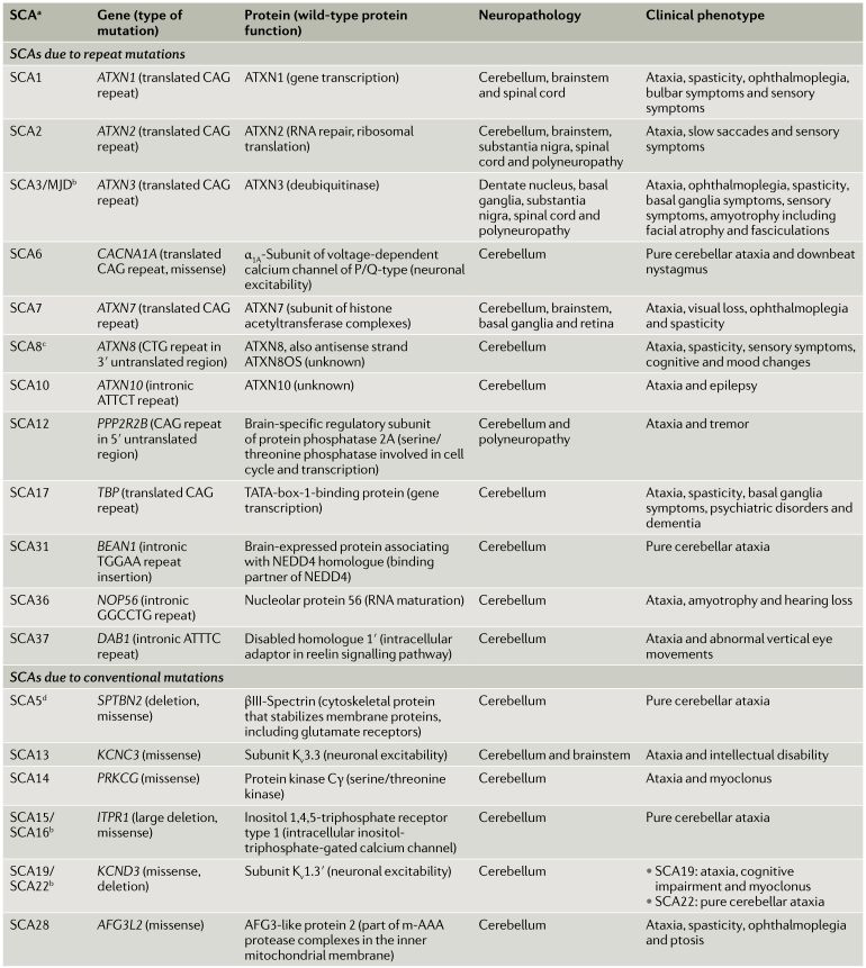
图2 SCA不同分型的神经病理学改变及其临床特点
SCA发病机制
SCA发病机制目前尚不清楚,主要与遗传因素有关,但也可能与以下几种因素相关。
1、蛋白毒性:蛋白毒性在多聚谷氨酰胺SCA的发病机制中具有中心作用,突变型蛋白容易构象异常并聚集,可招募正常的蛋白或分子伴侣在细胞内共同形成聚集体(更倾向于在核内形成),通过多种机制使细胞内的蛋白平衡破坏而产生不同的下游路径。另一些非重复扩展型SCA(如SCA14、SCA35)中的传统突变也可导致蛋白的错误折叠而异常聚集。
2、RNA毒性:对于非翻译区重复扩张型SCA,含有重复序列的转录本形成核内RNA团簇,这些团簇隔离RNA结合蛋白,干扰剪接或其他依赖RNA的过程,甚至影响细胞内蛋白平衡,导致细胞毒性。
3、离子通道功能障碍:编码离子通道或调节通道活性信号通路组件的基因致病性突变,会导致小脑神经电生理信号异常或驱动神经元功能障碍。
4、生物能量受损:突变型蛋白直接影响线粒体的结构和功能(SCA28),或如多聚谷氨酰胺SCA疾病蛋白介导的转录失调、自噬/溶酶体通路等细胞途径间接影响线粒体功能,从而影响生物能量产生。
5、核完整性丧失:核完整性丧失是指细胞核结构和功能均受到破坏,其中包括基因表达失调、DNA修复受损、核浆运输被破坏等。
6、其他:近年来,在重复扩张疾病中发现一种非ATG介导的RNA翻译,形成易于聚集的多肽,而这种翻译存在于一些SCA中,但RNA翻译在SCA中存在何种作用尚不明确。
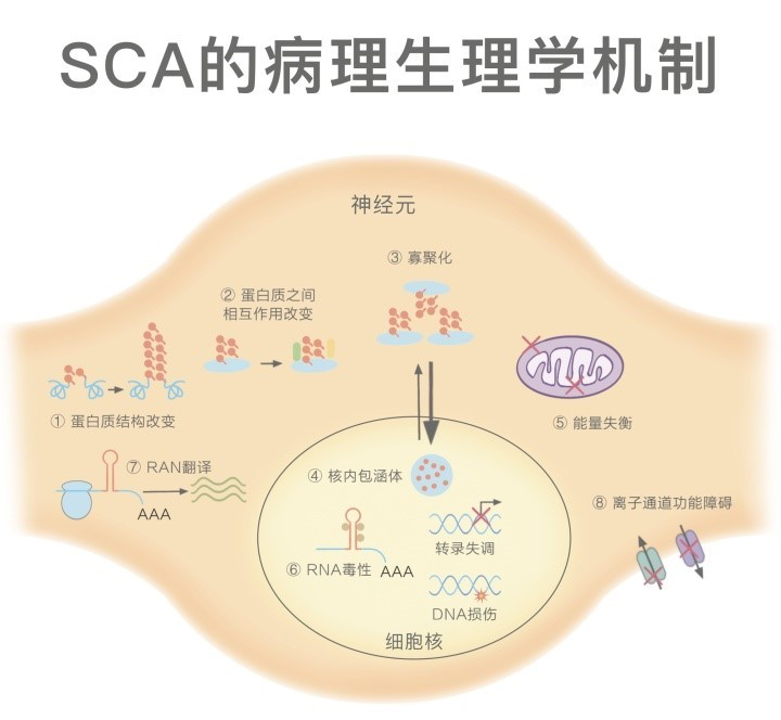
图3 SCAs的病理生理学机制
SCA病理改变
SCA共同的病理改变是小脑、脑干、脊髓变性和萎缩,往往选择性累及某一区域的神经元,且通常是对称性的改变,但其不同亚型也各有其特点。如SCA1主要是小脑、脑干的神经元丢失,脊髓小脑束和后索受损,很少累及黑质、基底核及脊髓前角细胞[4];SCA2以下橄榄核、脑桥、小脑损害为重;SCA3主要损害脑桥和脊髓小脑束;SCA7的特征是视网膜神经细胞变性。
1、大体所见:小脑萎缩,重量减轻,小脑沟回变宽;脑干变小,萎缩;脊髓萎缩,颈段及上胸段明显。
2、镜下所见:神经细胞脱失,小脑皮层的浦肯野细胞、颗粒细胞脱失,齿状核神经细胞脱失,小脑白质纤维及皮质脊髓束、脊髓小脑束、后索髓鞘脱失及轴索变性。橄榄小脑束、桥脑小脑束、脑桥横纤维等脱失及轴索变性。由于轴索增生,轴索球形成。
除上述神经系统改变外,少数病例还可见到内分泌、皮肤、骨骼等方面的改变,如:心肌肥厚、糖尿病、鱼鳞病和毛细血管扩张、脊柱侧弯和弓形足等。
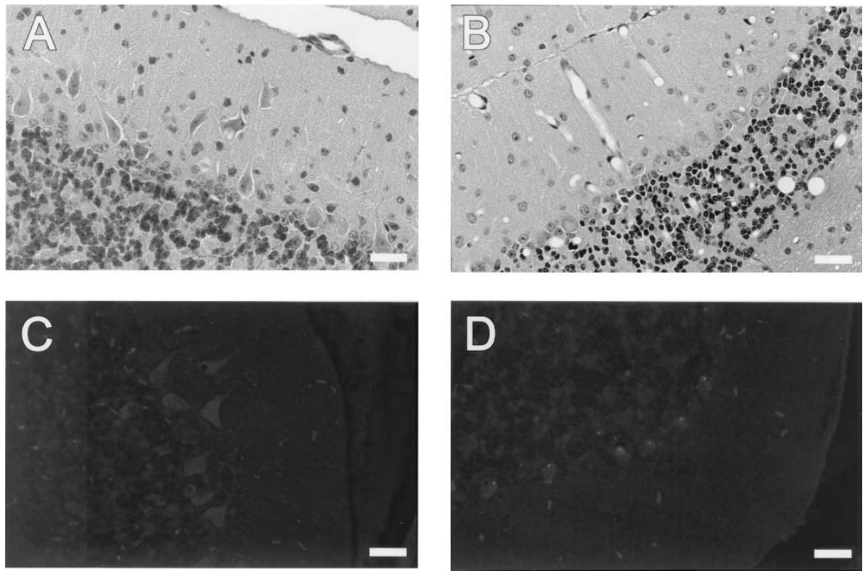
图4 苏木精和伊红染色(A和B)、泛素染色(C和D)12周龄小鼠小脑切片。(A)和(C) 12周脊髓小脑共济失调-1小鼠(SCA-1):Calbindin-null双突变体;(B)和(D) 12周龄的SCA-1杂合转基因小鼠。双突变体表现为Ataxin-1病理增加(A),伴有浦肯野细胞异位和浦肯野细胞层紊乱。在SCA-1无Calbindin双突变小鼠中,浦肯野细胞显示无可见包涵体的弥漫泛素免疫染色(C),而在年龄匹配的SCA-1杂合动物中(D),90%的浦肯野细胞含有泛素化的Ataxin-1核包涵体。比例:25μm
SCA临床表现
脊髓小脑性共济失调的临床表现各不相同。最常见的SCAs可影响中枢和神经系统的多个部位,小脑性共济失调是各型的共同特征,而其他区别性特征可能提示特定类型。除了小脑性共济失调外,可伴有眼球运动障碍、慢眼运动、视神经萎缩、视网膜色素变性、锥体束征、锥体外系征、肌萎缩、周围神经病和痴呆等。而部分SCA只引起小脑性共济失调。SCA初期的临床表现为走路不稳,肢体摇晃,行动反应迟缓以及反应性变差等。中期时的表现为说话时构音模糊不清,没有办法控制音调,眼球转动不流畅,看物时会出现“重影”等,同时也有肌肉不协调感加重,写字困难,有时也可出现吞咽困难,进食也容易出现呛咳等。到晚期时,说话困难,发音不清楚加重,肢体乏力,站立困难,无法行走,才需要靠轮椅等才能行走;理解力也逐步下降,最终发展到意识模糊,昏睡不清。
SCA诊断依据
SCA的诊断主要通过脑神经系统的临床检查程序来判断患者是否存在小脑及脊髓神经失调。同时也会询问患者的家族史,包括已故的亲人,通过核磁共振(MRI)以及基因检测来判断患者是否患有小脑萎缩症[5, 6]。脊髓小脑的早期诊断尤为重要,但是由于遗传的异质性,仍然无法根据临床症状对SCA的分型做出具体的诊断,基因检测仍然是SCA确诊的金标准。因此,明确SCA患者的基因诊断,并了解其发病机制,对进一步探索SCA具有非常重要的意义。
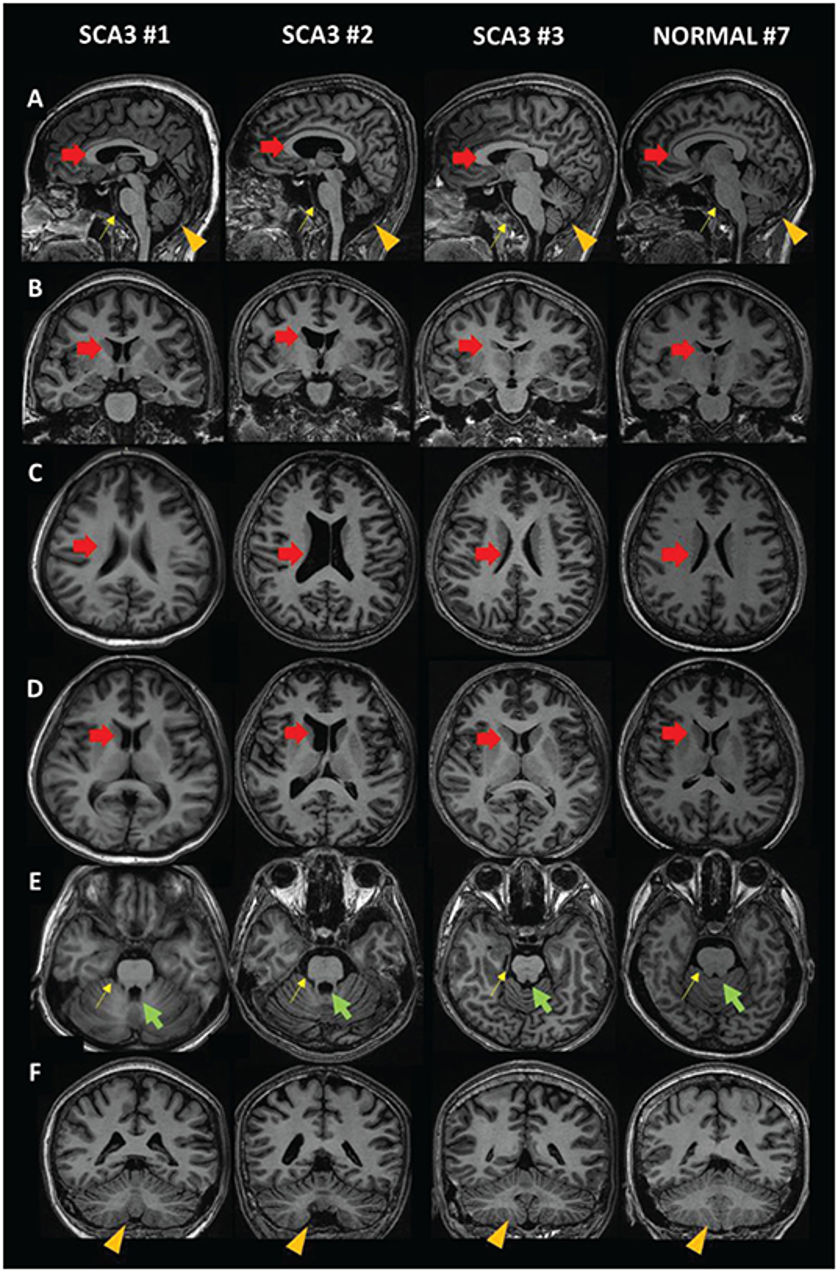
图5 SCA3患者与正常受试者的MRI图像对比。(A)矢状面。(B,F)冠状面。(C-E)轴向视图。红色箭头:侧脑室(SCA3扩张);绿色箭头:第四脑室(SCA3扩张);黄色箭头:脑干(SCA3中缩小);橙色箭头:小脑(SCA3尺寸减小)。
SCA治疗方法
目前SCA最新的治疗方法,主要集中在基因治疗和干细胞治疗两个方面。但这两个方面的研究或治疗,目前主要集中在临床试验或者是动物实验水平。目前尚无能有效阻止或减缓SCA进展的治疗方法,临床主要以对症支持治疗为主。SCA的治疗是涉及多学科的综合治疗,每种SCA都需要特定的治疗方案。
1、基因治疗:是指将人的正常基因或者有治疗作用的基因通过一定方式导入人体靶细胞,以纠正基因缺陷或者发挥治疗作用,从而达到治疗疾病的目的。然而目前这种方法还没有在临床上得到进一步的证实。
2、干细胞治疗:干细胞移植手术已在国内临床上相继开展,其基本原理为诱导多能干细胞分化为神经干细胞,替换受损细胞,并通过旁分泌作用为神经传导提供更好的微环境。但其有效性和安全性还有待于进一步观察研究。
参考文献:
[1].Duenas, A.M., R. Goold and P. Giunti, Molecular pathogenesis of spinocerebellar ataxias. Brain, 2006. 129(Pt 6): p. 1357-70.
[2].Durr, A., Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol, 2010. 9(9): p. 885-94.
[3].Corral-Juan, M., et al., Clinical, genetic and neuropathological characterization of spinocerebellar ataxia type 37. Brain, 2018. 141(7): p. 1981-1997.
[4].Vig, P.J., S.H. Subramony and D.O. McDaniel, Calcium homeostasis and spinocerebellar ataxia-1 (SCA-1). Brain Res Bull, 2001. 56(3-4): p. 221-5.
[5].Zheng F., et al., [Modification and evaluation of assessment of medication literacy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2016. 41(11): p. 1226-1231.
[6].Sobana, S.A., et al., Brain MRI Volumetry Analysis in an Indonesian Family of SCA 3 Patients: A Case-Based Study. Front Neurol, 2022. 13: p. 912592.
类型:原创
病例ID:ZYLM000004183
校对:陆喜红
阅读:1366
文章已于2024-01-15修改




