

男,18Y。
主诉:呼吸时胸痛半天,无咳嗽。
身高:183cm
体重:45kg
手指细长,四肢细长。
病案讨论
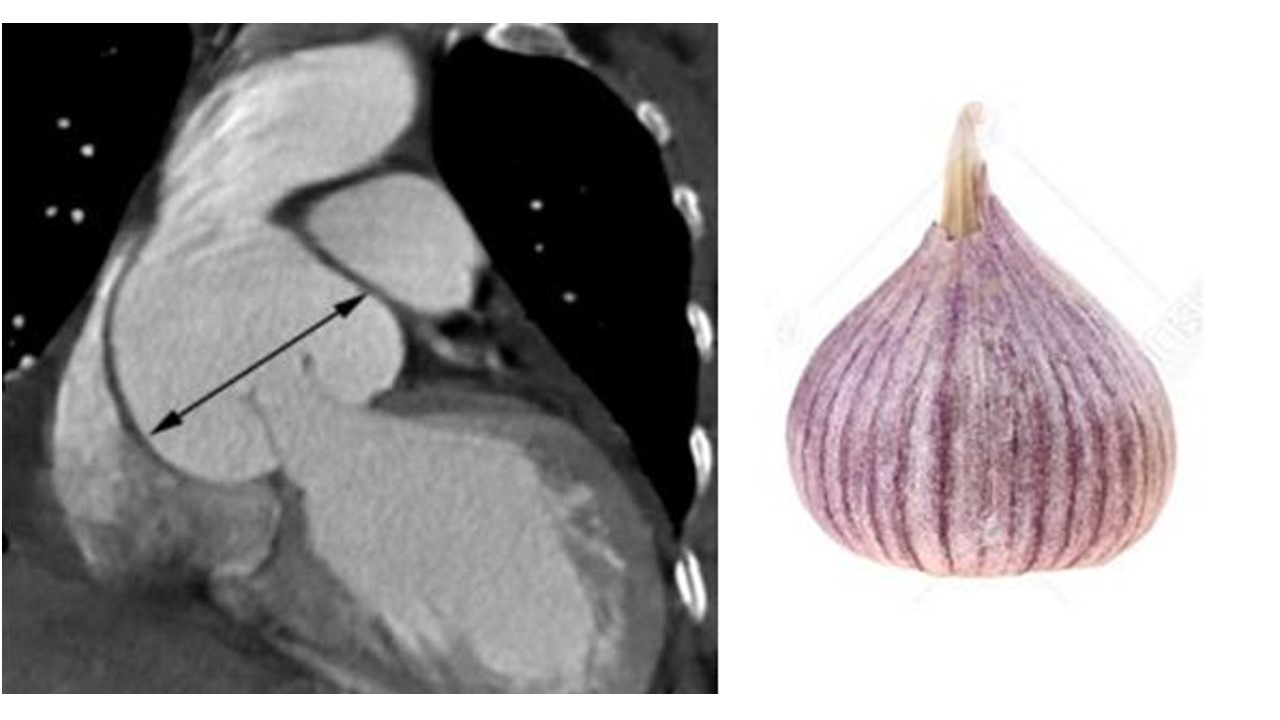
CT平扫:主动脉根部扩张形似蒜头。

马凡综合征(Marfan syndrome)亦称为 Marchesani 综合征、蜘蛛指(趾)征、肢体细长症等。
最初由法国小儿科医师安东尼·马凡(Antoine Marfan)于1896 年发现并描述,为一种以结缔组织为基本缺陷的遗传性疾病。该病为染色体显性遗传病,原纤维蛋白-1因子 (FBNI) 突变引起。患病率与种族、性别无关,人群中发病率为0.02%~0.03%。

马凡综合征的主要表现是骨骼、眼睛以及心血管三大系统的发育异常,其特征为患者身材高,四肢、手指、脚趾都很细长,称为蜘蛛指。眼部表现主要有晶体状脱位或半脱位、高度近视、白内障、视网膜剥离、虹膜震颤等。男性多于女性。在心血管方面,马凡综合征的患者大动脉血管中膜往往发育不良,非常薄弱,容易发生主动脉破裂。
病理基础:FBNI 编码大量的糖蛋白,是创建多数组织细胞外微纤维的主要成分。FBNI 突变引起微纤维异常导致弹性纤维稀疏、中断、粘液样变及囊性坏死。最终引起动脉瘤和夹层。
马凡综合征的主要危害是心血管病变,特别是合并的主动脉瘤,所以应尽早发现,及时治疗。根据临床表现,心血管、眼、骨骼改变三主征,结合家族史,即可诊断。 马凡综合征在临床上主要分为两型:三主征俱全者称完全型;仅二项者称不完全型。
主动脉影像:主动脉根部扩张形似蒜头;导致主动脉瓣中心性关闭不全而产生主动脉瓣返流。
鉴别诊断:白塞氏病(Behçet’s disease)白塞氏病,是一种慢性的非细菌性炎症性疾病,最常表现为口腔或者是生殖器部位的反复溃疡,而实际上,它引起的主要病变是全身各种血管的炎症,包括动脉和静脉。该病可发于任何年龄,但多集中于20-30岁,男女性染病的概率整体上没有差别,在中东以及地中海地区,男性的发病率要高于女性,而在包括中国在内的东亚地区,则是以女性病人居多。
白塞氏病的主要临床表现为:
口腔和生殖器反复发生的多发性溃疡
皮肤和关节慢行疼痛
眼部炎症,葡萄膜炎,视网膜脱落等,严重者甚至致盲
全身各个脏器问题,尤其是神经系统问题
大小血管炎,包括动脉和静脉,引起血栓栓塞,
升主动脉以及主动脉弓部的瘤样扩张

马凡综合征的治疗:目前,马凡综合征没有根治的方法,无论是药物治疗还是手术治疗,都只是对症治疗。
类型:原创
病例ID:ZYLM000004581
校对:王宇军
阅读:1327
文章已于2024-02-27修改




